
About Medicine
PDlasta® (pegfilgrastim) is a covalent conjugate of recombinant methionyl human G‑CSF (Filgrastim) and monomethoxypolyethylene glycol. Filgrastim is a water‑soluble 175 amino acid protein with a molecular weight of approximately 19 kilodaltons (kD). Filgrastim is obtained from the bacterial fermentation of a strain of Escherichia coli transformed with a genetically engineered plasmid containing the human G‑CSF gene. To produce pegfilgrastim, a 20 kD monomethoxypolyethylene glycol molecule is covalently bound to the N‑terminal methionyl residue of Filgrastim. The average molecular weight of pegfilgrastim is approximately 39 kD.
Pharmaceutical form
Prefilled syringes: 6mg pegfilgrastim in 0.6 mL.
Qualitative composition
Sterile solution, clear and colorless in prefilled syringe.
Method of administration
PDlasta® is injected subcutaneously. The injections should be given into the thigh, abdomen or upper arm.
Guidance to patients
Overdosage
If you think that you have used too much or too little PDlasta®, tell your doctor, nurse or pharmacist immediately, even if you have no signs of a problem.
What if you forget to use PDlasta®?
If you forget to give yourself a dose, have it as soon as you remember. Do not give yourself a double dose on the same day to make up for a forgotten dose. Keeping a diary will help to make sure you do not miss a dose.
Effect on Ability to Drive and Operate Machinery
No studies on the effects on the ability to drive and use machines have been performed.
Use
1. Patients with Cancer Receiving Myelosuppressive Chemotherapy
PDlasta is indicated to decrease the incidence of infection, as manifested by febrile neutropenia, in patients with non-myeloid malignancies receiving myelosuppressive anti-cancer drugs associated with a clinically significant incidence of febrile neutropenia.
2. Patients with Hematopoietic Subsyndrome of Acute Radiation Syndrome
PDlasta is indicated to increase survival in patients acutely exposed to myelosuppressive doses of radiation.
Dosing: Adult
1. Patient with Cancer Receiving Myelosuppressive Chemotherapy
The recommended dosage of PDlasta is a single subcutaneous injection of 6 mg administered once per chemotherapy cycle. Do not administer PDlasta between 14 days before and 24 hours after administration of cytotoxic chemotherapy.
2. Patients with Hematopoietic Subsyndrome of Acute Radiation Syndrome
The recommended dose of PDlasta is two doses, 6 mg each, administered subcutaneously one week apart. Administer the first dose as soon as possible after suspected or confirmed exposure to radiation levels greater than 2 gray (Gy). Administer the second dose one week after the first dose. Obtain a baseline complete blood count (CBC). Do not delay administration of PDlasta if a CBC is not readily available. Estimate a patient’s absorbed radiation dose (i.e., level of radiation exposure) based on information from public health authorities, biodosimetry if available, or clinical findings such as time to onset of vomiting or lymphocyte depletion kinetics.
Dosing: Pediatric
Pediatric weighing more than 45 Kg
1. Patients with Cancer Receiving Myelosuppressive Chemotherapy: Like Adults
2. Patients with Hematopoietic Subsyndrome of Acute Radiation Syndrome: Like Adults
Pediatric patients weighing less than 45 kg
Volume to Administer
PDlasta Dose
Body Weight
See below*
See below*
Less than 10 kg*
0.15 mL
1.5 mg
10 -20 kg
0.25 mL
2.5 mg
21 -30 kg
0.40 mL
4 mg
31 -44 kg
*For pediatric patients weighing less than 10 kg, administer 0.1 mg/kg (0.01 mL/kg) of PDlasta.
Patients with renal impairment
No dose change is recommended in patients with renal impairment, including those with end stage renal disease.
Contraindications
Hypersensitivity to the active substance or to any of the excipients. (You should not use PDlasta® if you are allergic to pegfilgrastim or filgrastim).
Warnings and precautions
- Due to the potential sensitivity of rapidly dividing myeloid cells to cytotoxic chemotherapy‚ PDlasta® should be administered at least 24 hours after administration of cytotoxic chemotherapy. In clinical trials, PDlasta® has been safely administered 14 days before chemotherapy.
- Limited clinical data suggest a comparable effect on time to recovery of severe neutropenia for pegfilgrastim to filgrastim in patients with de novo acute myeloid leukemia However, the long-term effects of PDlasta® have not been established in acute myeloid leukemia; therefore, it should be used with caution in this patient population.
- Granulocyte-colony stimulating factor can promote growth of myeloid cells in vitro and similar effects may be seen on some non-myeloid cells in vitro.
- The safety and efficacy of PDlasta® have not been investigated in patients with myelodysplastic syndrome, chronic myelogenous leukemia, and in patients with secondary Acute Myeloid Leukemia (AML); therefore, it should not be used in such patient Particular care should be taken to distinguish the diagnosis of blast transformation of chronic myeloid leukemia from acute myeloid leukemia.
- The safety and efficacy of PDlasta® administration in de novo AML patients aged < 55 years with cytogenetics have not been established.
- The safety and efficacy of PDlasta® have not been investigated in patients receiving high dose chemotherapy. This medicinal product should not be used to increase the dose of cytotoxic chemotherapy beyond established dosage regimen.
- Uncommon (≥1/1,000 to < 1/100) pulmonary adverse reactions, in particular interstitial pneumonia, have been reported after G-CSF administrative Patients with a recent history of pulmonary infiltrates or pneumonia may be at higher risk.
- The onset of pulmonary signs such as cough, fever, and dyspnea in association with radiological signs of pulmonary infiltrates, and deterioration in pulmonary function along with increased neutrophil count may be preliminary signs of Acute Respiratory Distress Syndrome (ARDS). In such circumstances PDlasta® should be discontinued at the discretion of the physician and the appropriate treatment given.
- Capillary leak syndrome has been reported after granulocyte-colony stimulating factor administration and is characterized by hypotension, hypoalbuminemia, edema and hemoconcentration Patients who develop symptoms of capillary leak syndrome should be closely monitored and receive standard symptomatic treatment, which may include a need for intensive care.
- Uncommon but generally asymptomatic cases of splenomegaly and uncommon cases of splenic rupture, including some fatal cases, have been reported following administration of pegfilgrastim. Therefore, spleen size should be carefully monitored (g. clinical examination, ultrasound). A diagnosis of splenic rupture should be considered in patients reporting left upper abdominal pain or shoulder tip pain.
- Treatment with PDlasta® alone does not preclude thrombocytopenia and anemia because full dose myelosuppressive chemotherapy is maintained on the prescribed schedule Regular monitoring of platelet count and hematocrit is recommended. Special care should be taken when administering single or combination chemotherapeutic agents which are known to cause severe thrombocytopenia.
- Sickle cell crises have been associated with the use of pegfilgrastim in patients with sickle cell trait or sickle cell disease. Therefore, physicians should use caution when prescribing PDlasta® in patients with sickle cell trait or sickle cell disease, should monitor appropriate clinical parameters and laboratory status and be attentive to the possible association of this medicine with splenic enlargement and vaso-occlusive crisis.
- White blood cell (WBC) counts of 100 x 109/L or greater have been observed in less than 1% of patients receiving PDlasta®. No adverse events directly attributable to this degree of leukocytosis have been reporte Such elevation in white blood cells is transient, typically seen 24 to 48 hours after administration and is consistent with the pharmacodynamic effects of this medicine. Consistent with the clinical effects and the potential for leukocytosis, a WBC count should be performed at regular intervals during therapy. If leukocyte counts exceed 50 x 109/L after the expected nadir, this medicine should be discontinued immediately.
- Hypersensitivity, including anaphylactic reactions, occurring on initial or subsequent treatment have been reported in patients treated with PDlasta®. Permanently discontinue PDlasta® in patients with clinically significant hypersensitivity. Do not administer PDlasta® to patients with a history of hypersensitivity to pegfilgrastim or filgrastim. If a serious allergic reaction occurs, appropriate therapy should be administered, with close patient follow-up over several days.
- As with all therapeutic proteins, there is a potential for immunogenicity. Rates of generation of antibodies against pegfilgrastim is generally low. Binding antibodies do occur as expected with all biologics; however, they have not been associated with neutralising activity at present.
- The safety and efficacy of PDlasta® for the mobilization of blood progenitor cells in patients or healthy donors has not been adequately evaluated.
- Increased hematopoietic activity of the bone marrow in response to growth factor therapy has been associated with transient positive bone imaging finding This should be considered when interpreting bone-imaging results.
- PDlasta® contains sorbitol. Patients with rare hereditary problems of fructose intolerance should not take this medicine.
- PDlasta® contains less than 1 mmol (23 mg) sodium per 6 mg dose, i.e. essentially ‘sodium-free’.
- In order to improve the traceability of granulocyte-colony stimulating factors (G-CSFs), the trade name of the administered product should be clearly recorded in the patient file.
- Delayed myelosuppression: Use has not been evaluated with patients receiving chemotherapy associated with delayed myelosuppression (eg, nitrosoureas, mitomycin).
- Pediatrics: The 6 mg fixed dose should not be used in infants, children, and adolescents weighing <45 kg.
- May potentially act as a growth factor for any tumor type, particularly myeloid malignancies; caution should be exercised when using in any malignancy with myeloid characteristics. Tumors of nonhematopoietic origin may have surface receptors for pegfilgrastim.
- This medicinal product must not be mixed with other medicinal products, particularly with sodium chloride solution.
Pregnancy
There are no or limited amount of data from the use of pegfilgrastim in pregnant women. Studies in animals have shown reproductive toxicity. PDlasta® is not recommended during pregnancy and in women of childbearing potential not using contraception.
Lactation
There is insufficient information on the excretion of PDlasta® metabolites in human milk; a risk to the newborns/infants cannot be excluded. A decision must be made whether to discontinue breast-feeding or to discontinue/abstain from PDlasta® therapy taking into account the benefit of breast-feeding for the child and the benefit of therapy for the woman.
Adverse effects
Very common (≥ 1/10)
- Headache
- Nausea
- Bone pain
- Musculoskeletal pain (myalgia, arthralgia, pain in extremity, back pain, musculo-skeletal pain and neck pain)
Common (≥ 1/100 to < 1/10)
- Thrombocytopenia
- Injection site reaction (including injection site pain)
Uncommon(≥ 1/1,000 to < 1/100)
- Sickle cell crisis
- Leukocytosis
- Splenomegaly
- Splenic rupture,
- Hypersensitivity reactions
- Anaphylaxis
- Elevations in uric acid
- Acute Respiratory Distress Syndrome
- Pulmonary adverse reactions (interstitial pneumonia, pulmonary edema, pulmonary infiltrates and pulmonary fibrosis)
- Sweet’s syndrome (acute febrile dermatosis)
- Cutaneous vasculitis
- Non-cardiac chest pain
- Elevations in lactate dehydrogenase and alkaline phosphatase
- Transient elevations in LFT’s for ALT or AST
Rare (≥ 1/10,000 to < 1/1,000)
Capillary leak syndrome
Drug interactions
5-fluorouracil
Concomitant administration of PDlasta® and 5-fluorouracil (5-FU) or other antimetabolites has been shown to potentiate myelosuppression.
Lithium
The potential for interaction with lithium, which also promotes the release of neutrophils, has not been specifically investigated. There is no evidence that such an interaction would be harmful.
Nitrosoureas
The safety and efficacy of PDlasta® have not been evaluated in patients receiving chemotherapy associated with delayed myelosuppression e.g., nitrosoureas.
Specific interaction or metabolism studies have not been performed; however, clinical trials have not indicated an interaction of PDlasta® with any other medicinal product
Monitoring Parameters
Complete blood count (with differential) and platelet count should be obtained prior to chemotherapy. Leukocytosis (white blood cell counts 100,000/mm 3) has been observed in <1% of patients receiving pegfilgrastim. Monitor platelets and hematocrit regularly. Evaluate fever, pulmonary infiltrates, and respiratory distress; evaluate for left upper abdominal pain, shoulder tip pain, or splenomegaly. Monitor for sickle cell crisis (in patients with sickle cell anemia).
Mechanism of Action
PDlasta® stimulates the production, maturation, and activation of neutrophils, pegfilgrastim activates neutrophils to increase both their migration and cytotoxicity. Pegfilgrastim has a prolonged duration of effect relative to filgrastim and a reduced renal clearance.
Pharmacokinetics
Half-life elimination
SubQ: Adults: 15-80 hours; Children (100 mcg/kg dose): ~20-30 hours (range: up to 68 hours)
Peak serum concentration
After a single subcutaneous dose of pegfilgrastim, the peak serum concentration of pegfilgrastim occurs at 16 to 120 hours after dosing and serum concentrations of pegfilgrastim are maintained during the period of neutropenia after myelosuppressive chemotherapy.
Elimination
Serum clearance of pegfilgrastim decreases with increasing dose. Pegfilgrastim appears to be mainly eliminated by neutrophil mediated clearance, which becomes saturated at higher doses. Consistent with a self-regulating clearance mechanism, the serum concentration of pegfilgrastim declines rapidly at the onset of neutrophil recovery.
Guidance for injection
- Before use, leave the PDlasta® syringe to stand until it reaches room temperature. This usually takes between 15 and 30 minutes. Do not remove the syringe’s needle cover while allowing it to reach room temperature.
- Only take one dose of PDlasta® from each syringe.
- PDlasta® is given alone and not mixed with other liquids for injection.
- Do not shake PDlasta® Prolonged vigorous shaking may damage the product. If the product has been shaken vigorously, don’t use it.
- Check the syringe, to make sure it is the right dose, has not passed its expiry date, is not damaged, and the liquid is clear and not frozen.
- Choose an injection site. Do not inject PDlasta® into an area that is tender, red, bruised, hard, or has scars or stretch marks. Recommended sites for injection are:
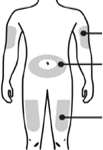
Upper part of your thigh.
Belly, except for a 5 cm (2-inch) area right around your belly button.
Outer area of upper arm (only if someone else is giving you the injection).
- Wash your hands. Use an antiseptic swab on the injection site, to disinfect it.
- Hold the pre-filled syringe by the body of the syringe with the covered needle pointing upward.
- Do not hold by the plunger head, plunger or needle cover.
- Do not pull back on the plunger at any time.
- Do not remove the needle cover from the pre-filled syringe until you are ready to inject your PDlasta®.
- Take the cover off the syringe by holding the barrel and pulling the cover off carefully without twisting it. Don’t push the plunger, touch the needle or shake the syringe.
- Pinch a fold of skin between your thumb and index finger. Don’t squeeze it.
- Push the needle in fully. Your doctor or nurse may have shown you how to do this.
- Push the plunger with your thumb as far as it will go to inject the entire amount of liquid. Push it slowly and evenly, keeping the skin fold pinched.
- When the plunger is pushed as far as it will go, take out the needle and let go of the skin.
- When the needle is pulled out of your skin, there may be a little bleeding at the injection site. This is normal. You can press an antiseptic swab over the injection site for a few seconds after the injection.
- Dispose of your used syringe in a safe container.
Storage and stability
- Store in a refrigerator (2°C – 8°C).
- Keep the container in the outer carton in order to protect from light.
- Keep out of the reach and sight of children.
- Do not use PDlasta® after the expiry date which is stated on the box and on the syringe label (EXP). The expiry date refers to the last day of that month.
- Do not freeze PDlasta® and do not use PDlasta® that has been frozen or improperly refrigerated.
- Do not shake PDlasta®.
- PDlasta® pre-filled syringes are single dose containers, discard any unused product.
Other composition
Sodium acetate, Sorbitol, Polysorbate 20 and Water for injections